
Quelle est la cause de l’amyotrophie spinale ?
L’amyotrophie spinale proximale liée à SMN1 (SMA ou amyotrophie spinale infantile, ASI) est due à des anomalies situées dans le gène SMN1, localisé sur le chromosome 5.
Ces anomalies entrainent l’absence de production de la protéine de survie des motoneurones SMN (pour survival of motor neuron), avec une perte de motoneurones, des cellules nerveuses qui commandent la contraction des muscles. Cela se traduit par une faiblesse et une fonte des muscles (amyotrophie) dit « proximaux », c’est-à-dire les plus proches du tronc : muscles des épaules et des bras pour les membres supérieurs et muscles des hanches et des cuisses pour les membres inférieurs.

Chez toutes les personnes atteintes de SMA, et chez 95% de la population générale, il existe en plus du gène SMN1 un autre gène quasiment identique, le gène SMN2. Cependant, ce gène SMN2 ne suffit pas à produire suffisamment de protéine SMN fonctionnelle.
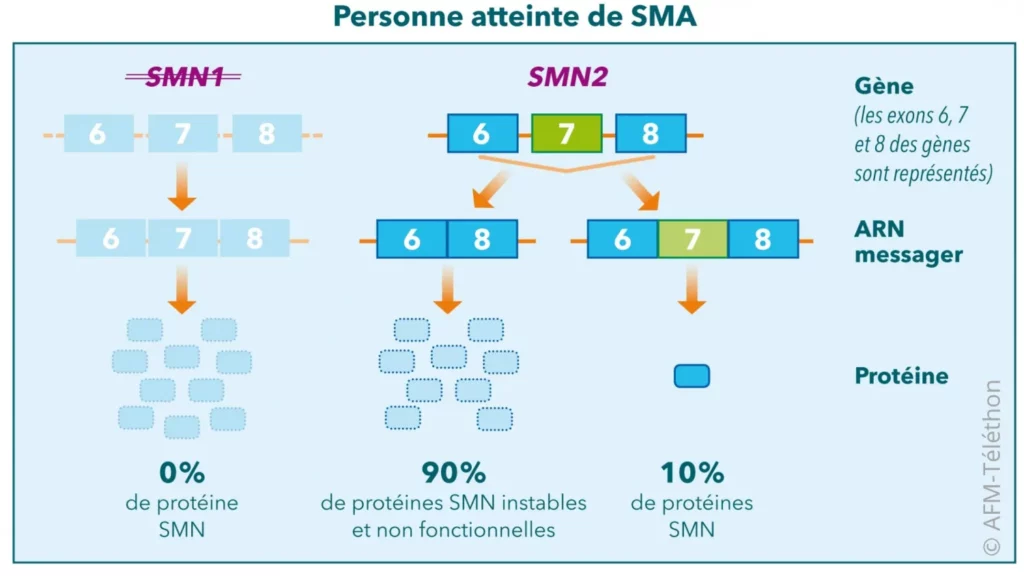
Les médicaments actuels et ceux à l’étude dans la SMA ciblent principalement les gènes SMN1 ou SMN2 dans le but d’augmenter la production de protéine SMN.
Les différents noms de la SMA
L’amyotrophie spinale proximale liée à SMN1 est aussi appelée amyotrophie spinale infantile (ASI), amyotrophie spinale 5q, maladie de la corne antérieure de la moelle épinière, maladie du motoneurone, amyotrophie spinale antérieure (ASA), ou, plus fréquemment, SMA (pour spinal muscular atrophy).
Dans quelles circonstances est posé le diagnostic ?
À la naissance, en l’absence de symptômes
L’arrivée des nouveaux traitements et la démonstration de l’intérêt de les utiliser précocement pour modifier l’évolution de l’amyotrophie spinale proximale liée à SMN1 (SMA), voire stopper sa progression, a poussé à développer son dépistage à la naissance dans plusieurs pays.
Le dépistage à la naissance de la SMA
Aussi appelé dépistage néonatal (DNN), il permet de savoir dès les premiers jours de vie, et le plus souvent avant même l’apparition des premiers symptômes, si un nouveau-né est porteur ou non d’anomalies génétiques de la SMA. Depuis quelques années, plusieurs régions ou pays du monde ont démontré que le dépistage néonatal de la SMA est faisable et efficace.
Désormais, aux États-Unis, dans tous les états, les nouveau-nés sont testés à la naissance pour la SMA.
Sur le continent européen, ce sont 72% des bébés qui sont dépistés pour la SMA à la naissance.
En France, le programme DEPISMA a démontré la faisabilité du dépistage néonatal de la SMA sur une durée de deux ans dans deux régions : la Nouvelle-Aquitaine et la région Grand Est.
Sa mise en place en France devrait se faire à partir du 1er septembre 2025.
Pour en savoir plus sur le programme DEPISMA
L’arrêté qui valide le dépistage néonatal généralisé pour l’ensemble du territoire français a été publié le 30 avril au journal officiel .
Avant la naissance, lorsque la maladie existe dans la famille
D’origine génétique, l’amyotrophie spinale proximale liée à SMN1 (SMA) est une maladie héréditaire, qui se transmet selon un mode autosomique récessif.
Si un membre d’une famille est atteint de SMA, d’autres membres de cette même famille peuvent avoir un risque de l’être aussi. Le recours au conseil génétique permet d’évaluer ce risque et d’accompagner une personne ou une famille confrontée à ce risque ou lors d’un projet d’enfant.
Lors de la grossesse, le diagnostic prénatal (ou anténatal) permet de déterminer si l’enfant à naître est porteur ou non d’anomalies génétiques en cause dans l’amyotrophie spinale proximale liée à SMN1.
Plus tardivement, en présence de symptômes
Aujourd’hui, le diagnostic de l’amyotrophie spinale proximale liée à SMN1 (SMA) est le plus souvent posé lors de l’apparition des premiers symptômes. Il s’agit principalement d’une faiblesse musculaire, d’importance variable, qui retentit sur les capacités motrices et la fonction respiratoire. Elle peut apparaître dès la naissance ou dans les tous premiers mois de vie, chez l’enfant, et plus rarement chez l’adolescent, voire à l’âge adulte.
La classification actuelle distingue quatre formes d’amyotrophie spinale proximale, selon l’âge de début de la maladie et la sévérité des symptômes. Il est généralement admis que plus les symptômes apparaissent tôt, plus l’évolution de la maladie est sévère.
- L’amyotrophie spinale de type I (SMA de type I ou maladie de Werdnig-Hoffmann) commence avant l’âge de 6 mois, parfois même dès la naissance. L’enfant n’est pas capable de s’assoir seul. Certains experts font aussi la distinction entre une forme très précoce de la maladie se manifestant avant la naissance par une diminution des mouvements du fœtus (forme dite type 0), la forme débutant entre la naissance et 3 mois (type I) et la forme débutant après 3 mois (type I bis). D’autres lui préfèrent une classification basée sur 3 lettres (A, B et C).
- L’amyotrophie spinale de type II (SMA de type II) a un début plus tardif, entre l’âge de 6 et 18 mois. L’enfant est capable de se tenir assis seul mais n’acquiert pas la marche.
- Dans l’amyotrophie spinale de type III (SMA de type III ou maladie de Kugelberg-Welander), les symptômes commencent après l’âge de 18 mois, généralement avant l’âge de 6 ans et évoluent lentement, sur de nombreuses années.
- Dans l’amyotrophie spinale de type IV (ou SMA de type IV), les symptômes débutent à l’âge adulte avec des difficultés à la marche. L’évolution est généralement très lente, voire absente dans certains cas.
La sévérité de la maladie semble également être étroitement liée au nombre de copies du gène SMN2 :
Lorsqu’une amyotrophie spinale (SMA) est suspectée, le médecin propose des examens complémentaires, comme le test génétique qui confirmera ou non le diagnostic de la maladie. Dans la SMA, il s’agit principalement de rechercher l’absence de copies du gène SMN1, en cause dans la maladie, et de déterminer le nombre de copies du gène SMN2. Ainsi, on peut distinguer les types de SMA.
La classification par type a été mise en œuvre avant l’arrivée des nouveaux traitements pour anticiper l’évolution de la maladie et préparer la prise en charge. Avec la mise en place des nouveaux traitements, l’évolution des patients varient fortement selon l’efficacité individuelle de ceux-ci.
A ce jour, la prise en charge est basée sur les capacités motrices du patient, ainsi les préconisations internationales sont basés sur trois types :
- « non sitter » / non assis : ne tenant pas assis de manière autonome
- « sitter » assis : pouvant tenir assis mais incapable de marcher
- « walker » ambulant : ayant la capacité de marcher au moins quelques pas en autonome.

Déjà trois médicaments
Longtemps sans traitement de « fond », c’est-à-dire qui ciblent la cause et non les conséquences de la maladie, l’amyotrophie spinale proximale liée à SMN1 (SMA) dispose désormais de trois traitements : le Spinraza®, le Zolgensma® et l’Evrysdi®.
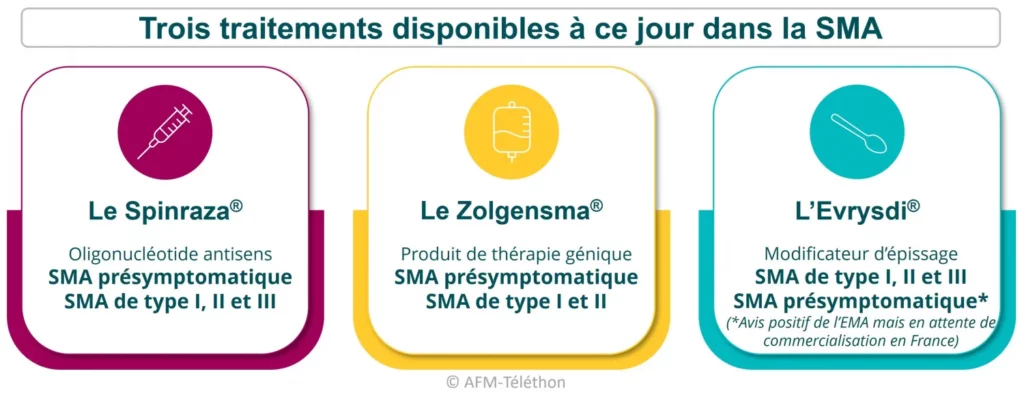
S’ils n’entrainent pas à une guérison complète de la maladie, ils améliorent le plus souvent considérablement la santé des patients et leur qualité de vie. Ils augmentent également leur espérance de vie.
Sans traitement : faiblesse musculaire, qui retentit sur les capacités motrices et la fonction respiratoire.
Avec un traitement après l’apparition des symptômes : effets bénéfiques sur le développement moteur, avec un recul de 6 ans environ (ces données pourront être adaptées sur le long terme).
Avec un traitement avant l’apparition des symptômes (via le dépistage néonatal) : développement normal dans la plupart des cas, avec un recul 4 ans environ (ces données pourront être adaptées sur le long terme).
Comment le type de traitement est-il décidé ?
La mise en place de ces traitements se fait au sein des consultations pluridisciplinaires spécialisées dans les maladies neuromusculaires.
Le Spinraza®
Commercialisé par le laboratoire Biogen, le Spinraza® (nusinersen) est un oligonulécotide antisens qui cible le gène SMN2 pour lui faire fabriquer la protéine SMN manquante.
Il permet sa réexpression dans les motoneurones avec un bénéfice clinique réel, notamment sur la fonction motrice, d’importance variable selon les personnes, le type de SMA, l’âge de début du traitement…
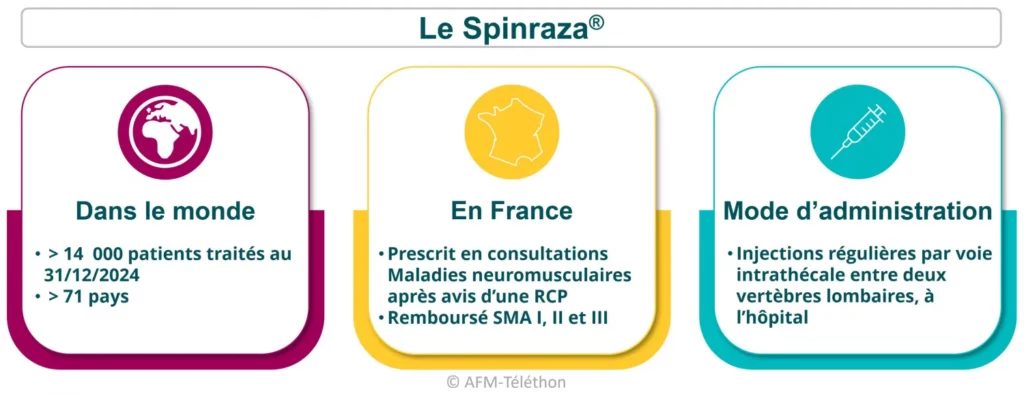
Des essais cliniques sont toujours en cours pour continuer à évaluer les effets du Spinraza.
Pour en savoir plus, voir la page « Le Spinraza® dans la SMA ».
Le Zolgensma®
Le Zolgensma® (onasemnogene abeparvovec) est un produit de thérapie génique qui vient suppléer le gène SMN1 défectueux par un gène SMN1 thérapeutique afin de fabriquer la protéine SMN, manquante dans la maladie.
Commercialisé par Novartis, il a montré des effets positifs et durables dans le temps : amélioration des performances motrices et respiratoires, allongement de l’espérance de vie… Les bénéfices sont cependant variables d’une personne à l’autre.

Plusieurs essais cliniques continuent d’évaluer ses effets dans différentes formes de SMA (type I, II, stade présymptomatique…). En France, plusieurs études sont en cours : l’essai STRENGTH, l’essai SPECTRUM, une étude de suivi à long terme du Zolgensma®.
Pour en savoir plus, voir la page « Le Zolgensma® dans la SMA »
L’Evrysdi®
Développé par le laboratoire Roche, l’Evrysdi® (risdiplam) est un modificateur d’épissage qui augmente la quantité de protéine SMN en corrigeant la maturation du gène SMN2. Il améliore la fonction motrice avec une efficacité durable dans le temps, mais variable selon les personnes.

Plusieurs essais sont en cours pour évaluer les effets de l’Evrysdi®, dont l’un en France : l’essai JEWELFISH, chez des patients âgés de 6 mois à 60 ans et ayant déjà été traités par Spinraza®, olésoxime ou Zolgensma®.
Pour en savoir plus, voir la page « L’Evrysdi® dans la SMA »
D’autres formes d’amyotrophie spinale
Différentes formes d’amyotrophie spinale proximale
- L’amyotrophie spinale avec hypoplasie ponto-cérébelleuse a été décrite en 1961. Elle se manifeste dans les 6 premiers mois de vie par une faiblesse musculaire et une hypotonie (amyotrophie spinale) auxquelles vont se rajouter des difficultés de coordination (atteinte cérébelleuse). Elle se transmet sur le mode autosomique dominant. L’anomalie génétique en cause a été découverte en 2009. Elle implique le gène VRK1, qui code une enzyme essentielle à la formation du noyau cellulaire.
- L’amyotrophie spinale récessive liée à l’X avec arthrogrypose a été décrite en 1991. Elle se manifeste dès la naissance par une hypotonie et une faiblesse musculaire (amyotrophie spinale) associées à une ankylose de certaines articulations (arthrogrypose). Elle se transmet sur le mode récessif lié au chromosome X. Elle est due à des anomalies découvertes en 2008 dans le gène UBE1, ce qui entraine un dysfonctionnement dans la dégradation et le recyclage des protéines (voie ubiquitine-protéasome).
- Il existe d’autres formes telles que celle prédominant aux membres inférieurs (SMA-LED) liée à des anomalies des gènes DYNC1H1 et BICD2, celle avec épilepsie myoclonique progressive (SMA-PME) liée à des anomalies dans le gène ASAH1, la maladie de la corne antérieure de la moelle avec arthrogrypose léthale (LAAHD) liée à des anomalies dans le gène GLE1…
Les amyotrophies spinales distales
Il s’agit de maladies rares, d’origine génétique, qui touchent les cellules nerveuses qui commandent les muscles. Elles sont dites « distales » car l’atteinte musculaire débute par celle des muscles des extrémités des membres (muscles distaux) : muscles des pieds et des jambes pour les membres inférieurs, muscles des mains et des avant-bras pour les membres supérieurs.
La majorité des amyotrophies spinales distales se manifeste par une faiblesse musculaire des muscles de la jambe et de la cheville. C’est pourquoi les médecins parlent aussi d’amyotrophies tibio-péronières.
Moins souvent ces maladies touchent préférentiellement les mains et les avant-bras, ou encore prédominent aux muscles des épaules et des chevilles (amyotrophie scapulo-péronière).
Dans certaines formes d’amyotrophie spinale distale, il peut y avoir une atteinte des muscles respiratoires (diaphragme) ou des cordes vocales (voix rauque…).
L’amyotrophie bulbo-spinale
Une amyotrophie bulbo-spinale est une maladie d’origine génétique, très rare, qui touche les cellules nerveuses commandant les mouvements : les motoneurones. Les muscles des jambes et certains muscles du visage et de la gorge (muscles dits ‘bulbaires », qui ont donné leur nom à ces formes d’amyotrophie spinale) sont moins forts.
Il existe plusieurs formes d’amyotrophie bulbo-spinale :
- la maladie de Kennedy, qui débute plutôt à l’âge adulte et touche les hommes (elle est liée au chromosome X)
- les amyotrophies bulbo-spinales de l’enfant : la maladie de Fazio-Londe (également appelée « paralysie bulbaire progressive de l’enfant ») et le syndrome de Brown-Vialetto-van Laere, qui apparaissent dès l’enfance, chez les filles comme chez les garçons. Le syndrome de Brown-Vialetto-van Laere touche par exemple 3 filles pour 1 garçon.






