

SMN
La protéine SMN
L’amyotrophie spinale est une maladie génétique, c’est-à-dire qu’elle est due à l’altération (mutation) ou à l’absence (délétion) d’un gène. Il s’agit du gène nommé SMN. En fait, ce gène défectueux ou absent n’est pas capable de donner les bonnes informations pour produire une protéine appelée du même nom SMN (protéine de « survie du motoneurone »). La protéine SMN est par conséquent déficiente, et le bon fonctionnement des motoneurones, ou neurones moteurs, est alors impossible. Les motoneurones sont des cellules nerveuses transmettant les ordres de mouvement entre la moelle épinière et les muscles qui vont effectuer le mouvement commandé. Sans la protéine SMN, les motoneurones permettant de « donner des ordres » aux muscles sont lésés et meurent. Les muscles en question deviennent inactifs, s’affaiblissent et s’atrophient.
Le gène SMN1
La plupart des gens possèdent deux exemplaires du gène survival motor neuron 1 (SMN1) : chacun est hérité d’un des parents. Ces gènes contiennent les informations nécessaires à la fabrication de la protéine SMN laquelle permet de conserver en vie les motoneurones périphériques. Les personnes atteintes d’amyotrophie spinale 5q ont une anomalie ou une mutation sur les deux exemplaires de leur gène SMN1, ce qui signifie qu’ils ne peuvent pas produire la protéine SMN dont ils ont besoin. Sans elle, les motoneurones périphériques se dégradent, ce qui restreint leur capacité à transmettre les signaux électriques du cerveau pour mouvoir les muscles altérés par l’amyotrophie spinale.
Le gène SMN1 se situe sur le chromosome 5 dans la région appelée « q », raison pour laquelle les principaux types d’amyotrophie spinale sont souvent nommés SMA 5q.
Le gène SMN2
Un autre gène joue un rôle dans la production de la protéine SMN : il s’agit du gène survival motor neuron 2 (SMN2), parfois appelé « gène de secours » de l’amyotrophie spinale. Cependant, une grande partie de la protéine SMN produite par SMN2 ne dispose pas de la composante essentielle habituellement produite par SMN1. Autrement dit, chez les personnes atteintes d’amyotrophie spinale, SMN2 peut produire une certaine quantité de protéine SMN mais pas compenser totalement l’altération du gène SMN1. Le nombre de copies de SMN2 (qui peut aller de zéro à huit) varie d’une personne à l’autre. Un nombre élevé de copies du gène SMN2 est généralement associé à des symptômes atténués. Le tableau 2 indique le nombre généralement attendu de copies de SMN2 associé à chaque type d’amyotrophie spinaleIl est toutefois impossible de prévoir précisément la gravité de l’amyotrophie spinale uniquement à partir de ce nombre de copies. Cela s’explique en partie par d’autres facteurs génétiques qui affectent la quantité de protéine SMN produite par le(s) gène(s) SMN2.
Tableau 2. Le nombre de copies de SMN2 généralement associé au type de SMA.
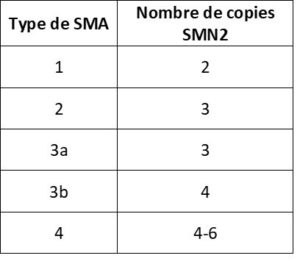
Note : Le nombre de copie en fonction du type est le nombre qui statistiquement est le plus fréquent, mais de nombreux patients ont pour un type donné un nombre de copies différent de celui généralement attendu.
Transmission de la SMA
L’amyotrophie spinale infantile se transmet des parents aux enfants par les anomalies des gènes SMN1. En règle générale, il s’agit d’un mode de transmission autosomique récessif, ce qui signifie que les anomalies des gènes SMN1 sont transmises des parents aux enfants. Autrement dit :
- Les personnes qui héritent d’une anomalie sur les deux exemplaires du gène SMN1 (un de chaque parent) sont atteintes d’amyotrophie spinale.
- Les personnes qui héritent d’une anomalie sur un exemplaire et qui ont un exemplaire sain du gène SMN1 (un de chaque parent) sont dits porteuses de l’amyotrophie spinale. Environ 1 personne sur 40 est porteuse de l’amyotrophie spinale. Les personnes porteuses ne sont pas atteintes par la maladie et n’ont aucun symptôme.
- Les personnes qui héritent de deux exemplaires sains du gène SMN1 (un de chaque parent) ne sont pas atteintes d’amyotrophie spinale et ne sont pas porteuses.
Bien que la plupart des personnes atteintes héritent de l’amyotrophie spinale par leurs deux parents, environ 2 % d’entre elles le sont par une mutation nouvelle, très probablement due à une erreur dans l’ADN de l’ovule ou du spermatozoïde desquels elles ont été conçues. Il s’agit d’une mutation dite de novo.
Il est important que les membres de la famille comprennent les résultats des tests génétiques ayant abouti au diagnostic d’amyotrophie spinale. Le conseil génétique doit avoir lieu avec un généticien ou un conseiller en génétique. Il répondra à toutes les questions que la famille se pose sur les aspects génétiques du diagnostic et fournira des renseignements sur les options possibles en cas de futures grossesses.
Les schémas suivants expliquent les risques, pour chaque grossesse, d’avoir un enfant atteint d’amyotrophie spinale dans différentes situations familiales. Les risques sont les mêmes que l’on soit du sexe masculin ou féminin. Le genre des parents et des enfants représentés dans les schémas sont à visée purement illustrative.
Famille 1. Les deux parents sont porteurs.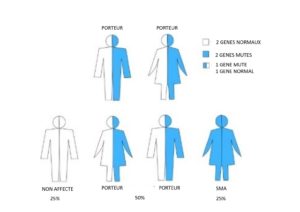
Pour chaque grossesse, il y a :
- 25 % de risque (1 sur 4) que l’enfant hérite d’une anomalie sur les deux exemplaires du gène SMN1 et soit atteint d’amyotrophie spinale
- 50 % de risque (1 sur 2) que l’enfant hérite d’une anomalie sur un exemplaire et d’un exemplaire sain du gène SMN1 et soit porteur
- 25 % de chance (1 sur 4) que l’enfant hérite de deux exemplaires sains du gène SMN1 et ne sera ni porteur, ni atteint d’amyotrophie spinale
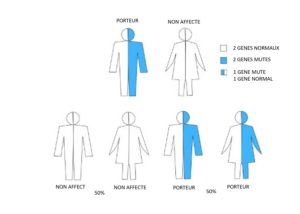
Famille 2. Un parent est porteur ; l’autre parent n’est pas atteint d’amyotrophie spinale et n’est pas porteur.
Pour chaque grossesse, il y a :
- 50 % de chance (1 sur 2) que l’enfant hérite de deux exemplaires sains du gène SMN1 et ne sera ni porteur, ni atteint d’amyotrophie spinale
- 50 % de risque (1 sur 2) que l’enfant hérite d’une anomalie sur un exemplaire et d’un exemplaire sain du gène SMN1 et soit porteur
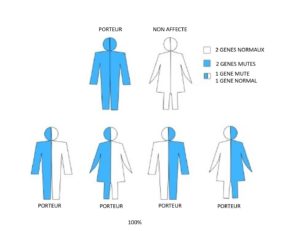
Famille 3. Un parent est atteint d’amyotrophie spinale ; l’autre parent n’est pas atteint d’amyotrophie spinale et n’est pas porteur.
Pour chaque grossesse, il y a :
- 100 % de risque (4 sur 4) que l’enfant hérite d’une anomalie sur un exemplaire et d’un exemplaire sain du gène SMN1 et soit porteur
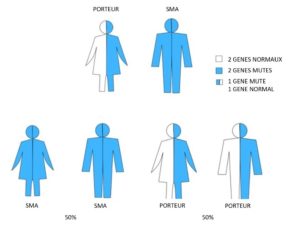 Famille 4. Un parent est atteint d’amyotrophie spinale ; l’autre parent est porteur.
Famille 4. Un parent est atteint d’amyotrophie spinale ; l’autre parent est porteur.
Pour chaque grossesse, il y a :
- 50% de risque (1 sur 2) que l’enfant hérite d’une anomalie sur les deux exemplaires du gène SMN1 et soit atteint d’amyotrophie spinale
- 50% de risque (1 sur 2) que l’enfant hérite d’une anomalie sur un exemplaire et d’un exemplaire sain du gène SMN1 et soit porteur
Famille 5. Les deux parents sont atteints d’amyotrophie spinale.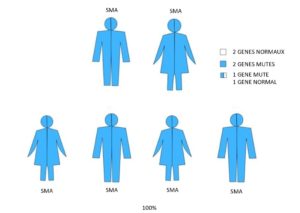
Pour chaque grossesse, il y a :
- 100 % de risque (4 sur 4) que l’enfant hérite d’une anomalie sur les deux exemplaires du gène SMN1 et soit atteint d’amyotrophie spinale
Sources :
- TREAT NMD Family guide
- L’amyotrophie spinale proximale : Encyclopédie Orphanet Grand Public







J’ai apris beaucoup d’information intéressante sur la SMA.
C’est génial